Em virtude de, estar acompanhando meu sobrinho em tratamento de saúde, as postagens estão adiadas por intervenção e força do momento.
Grato por toda compreensão, e as mensagens de apoio recebidas.
José Carlos
segunda-feira, 22 de outubro de 2012
quarta-feira, 3 de outubro de 2012
Diagnóstico Precoce
POSTAGEM DE GRANDE IMPORTÂNCIA
O Programa Nacional de Diagnóstico Precoce iniciou-se em 1979, por iniciativa do Instituto de Genética Médica, incluindo inicialmente apenas o rastreio da Fenilcetonúria (PKU).
Atualmente esta lista é bem mais extensa, e
o programa tende a crescer.
Esta postagem, é uma parcela do que há na página específica de mesmo nome Programa Nacional de Diagnóstico Precoce.
Lá estão detalhadas de forma específica as doenças investigadas, links importantes, redirecionamento para centros de diagnósticos, investigação, tratamentos, área científica e outros.
Contudo, há como seguir diretamente ao website:
Lá estão detalhadas de forma específica as doenças investigadas, links importantes, redirecionamento para centros de diagnósticos, investigação, tratamentos, área científica e outros.
Contudo, há como seguir diretamente ao website:
- Teste do pezinho
(entenda) Resultado
|
|
|
terça-feira, 2 de outubro de 2012
Acatalassemia
Histórico
Acatalassemia foi descrita no Japão por
Takahara e Miyamoto (1948), onde observaram que em casos de gangrena oral
progressiva, o peróxido de hidrogênio aplicado nas áreas ulceradas
não produzia espumas, como o usual. Também foi detectada na Suíça (Aebi
et al.,1962) e em Israel (Szeinberg et al., 1963).
Acatalassemia é uma desordem congênita decorrente
de uma deficiência de catalase eritrocitária, uma enzima responsável pela
quebra do peróxido de hidrogênio.
A doença é muito rara na população em geral, com
uma prevalência estimada de 1 em 31.250.
A desordem é geralmente assintomática, mas pode
estar associada com ulcerações orais e gangrena, ou diabetes mellitus e
aterosclerose em determinadas populações.
A transmissão é autossômica recessiva.
Distúrbios Peroxissômicos são um grupo de doenças
metabólicas geneticamente heterogêneas que a disfunção partes de peroxissomos. Os
peroxissomas são organelas celulares que desempenham uma parte integrante da
via metabólica. Medem cerca de 0,5 m de diâmetro e pode variar em tamanho entre
as diferentes espécies. Eles participam em importantes percursos
metabólicos específicos, tais como a beta-oxidação de muito longa cadeia de
ácidos gordos (VLCFA) e desintoxicação de peróxido de hidrogénio. Peroxissomas
também estão envolvidos na produção de colesterol, ácidos biliares, e
plasmalogens, que contribuem para uma grande parte do conteúdo de fosfolípide
da matéria cerebral branca.
Zellweger descreveu o primeiro caso de desordem
peroxissomal. Ao longo dos próximos três anos, uma série de relatos de
casos seguidos. A descrição inicial do peroxissomo apareceu em 1954 em uma
tese de doutorado sobre os rins de ratos, quase 10 anos após a descrição do primeiro
caso da doença peroxissomal foi publicado. Um estudo realizado em 1979, de
iniciar reações em sínteses de lipídios complexos em peroxissomas de fígado de
rato foi conduzido. Seus resultados ajudaram os investigadores a
compreender o papel destas organelas na doença humana.
Variantes
Suíça
Consiste de uma mutação na parte do gene que
codifica a estrutura, resultando na produção de uma catalase instável. Esta
catalase imperfeita facilita as infecções por bactérias que geram
peróxidos, como os estreptococos e os pneumococos, já que a catalase se vê
impotente para proteger os tecidos frente aos ataques deste tipo de bactérias.
Também pode ocorrer um acúmulo de peróxidos bacterianos nas zonas infectadas,
ocasionando uma disfunção nos neutrófilos. Esta variante geralmente é benigna e
assintomática ou com poucos sintomas.
Japonesa
Também é conhecida como enfermidade de Takahara.
Ocorre devido a uma mutação na parte reguladora do gene, que resulta na síntese
de enzima diminuída ou com menor atividade enzimática. As manifestações
clínicas da enfermidade de Takahara são infecções de boca (gengiva e amídalas)
com ulcerações e gangrena, que podem ocasionar uma enfermidade
periodontal prematura.
Diagnóstico
O diagnóstico se confirma mediante exames de
laboratório: a atividade da catalase nos eritrócitos, aparece muito reduzida e
o sangue em contato com água oxigenada torna-se marrom, e não produz as típicas
bolhas de oxigênio.
Tratamento
O tratamento consiste exclusivamente na
erradicação de possíveis infecções, e em intervenções periodontais adequadas
para evitá-las. Considera-se esta enfermidade como benigna.
Referências:
http://emedicine.medscape.com/article/1177387-overview
http://saude.psicologiananet.com.br/?s=Acatalassemia
Wikipédia, a enciclopédia livre.
sábado, 29 de setembro de 2012
Pseudo-adrenoleucodistrofia
SUMÁRIO
A deficiência da acil-CoA oxidase
peroxisomal é uma doença neurodegenerativa rara que pertence ao grupo das
doenças peroxisomais hereditárias e é caracterizada por hipotonia e convulsões
no período neonatal e regressão neurológica no início da infância.
A deficiência da acil-CoA oxidase
é uma doença rara com apenas 30-40 doentes identificados mundialmente até ao
momento.
A doença manifesta-se no período
neonatal com hipotonia (92%) e convulsões (91%) como características
dominantes. Pode estar presente dismorfismo facial (50%) com hipertelorismo,
epicanto, ponte nasal baixa, e baixa implantação das orelhas. Algumas crianças
têm polidactilia e hepatomegalia. O desenvolvimento psicomotor é atrasado, mas
as crianças são normalmente capazes de andar e dizer algumas palavras. Contudo,
a regressão neurológica ocorre normalmente aos 1-3 anos (idade média: 28
meses). A hipotonia é substituída por hipertonia com hiperreflexia. A epilepsia
pode tornar-se mais grave e pode surgir surdez neurossensorial. Pode ocorrer estrabismo,
nistagmo e atrofia ótica.
A deficiência da acil-CoA oxidase
peroxisomal é causada por mutações no gene ACOX1 (17q25.1) que codifica a
oxidase de cadeia lisa da acil-CoAperoxissomal. A transmissão é autossómica
recessiva.
O diagnóstico é baseado em
estudos laboratoriais revelando ácidos gordos de cadeia muito longa aumentados
(VLCFA) e atividade da acil-CoA oxidase marcadamente reduzida nos fibroblastos.
A RMN cerebral mostra sinais anormais da substância branca. O diagnóstico é
confirmado pela presença de mutações no gene ACOX1.
Os diagnósticos diferenciais
incluem a síndrome de Usher (ver este termo) e todas as causas de hipotonia
neonatal. As outras doenças peroxisomais devem ser descartadas, especialmente
adrenoleucodistrofia neonatal (ver este termo), que apresenta manifestações
clínicas semelhantes.
O diagnóstico antenatal é
possível através da análise bioquímica e/ou molecular dos amniócitos ou das células
das vilosidades coriónicas.
Deve ser oferecido aconselhamento
genético às famílias dos doentes.
Não está disponível tratamento
específico. Devem ser oferecidos cuidados de apoio multidisciplinares.
O prognóstico não é favorável; a
morte ocorre por volta dos 5 anos por problemas respiratórios.
Referências:
Pseudo-adrenoleucodistrofia
|
|
Editor(es)
Prof R.J.A. [Ronald] WANDERS
Atualizado em: Fevereiro 2010
sexta-feira, 28 de setembro de 2012
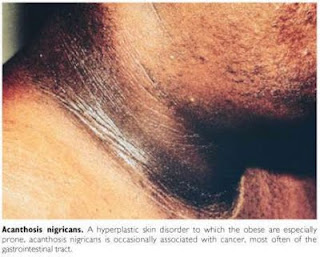
Acanthosis Nigricans (AN)
 A Acanthosis
nigricans (AN) é condição dermatológica caracterizada por espessamento,
hiperpigmentação e acentuação das linhas da pele, gerando aspecto grosseiro e
aveludado no local afetado. Histologicamente, é comum a observação de
hiperceratose, projeção acentuada das papilas da derme e discreto espessamento
das camadas da epiderme.1 Embora possa ocorrer em qualquer local da
superfície corpórea, a área mais atingida é a região posterior do pescoço,
seguida pelas axilas, face lateral do pescoço, superfícies flexoras dos
membros, região periumbilical, inframamária, mucosa oral ou mesmo, em casos
raros, planta dos pés e palma das mãos.
A Acanthosis
nigricans (AN) é condição dermatológica caracterizada por espessamento,
hiperpigmentação e acentuação das linhas da pele, gerando aspecto grosseiro e
aveludado no local afetado. Histologicamente, é comum a observação de
hiperceratose, projeção acentuada das papilas da derme e discreto espessamento
das camadas da epiderme.1 Embora possa ocorrer em qualquer local da
superfície corpórea, a área mais atingida é a região posterior do pescoço,
seguida pelas axilas, face lateral do pescoço, superfícies flexoras dos
membros, região periumbilical, inframamária, mucosa oral ou mesmo, em casos
raros, planta dos pés e palma das mãos. Basicamente,
AN pode ser dividida nas formas maligna e benigna. A forma maligna representa
um marcador de neoplasias abdominais, particularmente o adenocarcinoma
gástrico. As formas benignas são divididas em idiopática, hereditária, induzida
por drogas e as causadas por doenças endócrinas.
Basicamente,
AN pode ser dividida nas formas maligna e benigna. A forma maligna representa
um marcador de neoplasias abdominais, particularmente o adenocarcinoma
gástrico. As formas benignas são divididas em idiopática, hereditária, induzida
por drogas e as causadas por doenças endócrinas.
As
endocrinopatias são as principais causas de AN, sendo a obesidade o distúrbio
mais comum, freqüentemente associado ao hiperinsulinismo, ao diabetes
mellitus e à resistência à insulina. Outros distúrbios endócrinos
associados à AN são descritos: doença de Cushing, ovários policísticos,
tireoideopatias, hirsutismo, doença de Addison, acromegalia, entre outros,
alguns dos quais cursam com resistência à insulina.
A
prevalência de AN em populações não selecionadas varia de sete a 74%, de acordo
com idade, raça, freqüência do tipo e do grau de obesidade e da concomitância com
endocrinopatia. Em 34 indivíduos obesos de ambos os sexos, numa população
predominantemente de negros (59%), Hud e col. descreveram prevalência de
AN em 74% dos pacientes.
A
detecção e algumas dicas de saúde ganharam importância nos últimos
anos, embora acanthosis nigricans seja um problema que só causa transtornos
para a estética do paciente, sua origem está relacionada a distúrbios internos
responsáveis por doenças do aparelho circulatório, que em estágios avançados,
pode causar o entupimento ou ruptura das artérias, resultando em ataques do
coração e cérebro.
Embora
Acantose nigricans seja um problema provocado por problemas de constrangimentos
cosméticos, sua origem está associada também a distúrbios internos ou a doenças
cardiovasculares responsáveis por cânceres internos.
An. Bras. Dermatol. vol.77 no.5 Rio de
Janeiro Sept./Oct. 2002
http://www.saudedicas.com.br
quinta-feira, 27 de setembro de 2012
Acalvaria
Acalvaria
Uma rara condição congênita onde a calota craniana está faltando,
mas o resto do rosto e base do crânio é normal. A pele do couro cabeludo
simplesmente cobre o cérebro sem osso do crânio de proteção por baixo. Acalvaria
é um defeito congênito de causa desconhecida.
Os sintomas
Ausência de ossos chatos de calota craniana
Ausência de duramater e músculos associados
Anomalias crânio
Ausência de calota craniana
Condições médicas relacionadas
Para investigar as causas da acalvaria, considere pesquisar doenças
que podem ser semelhantes, ou associadas com acalvaria, segue-se o fluxo de
eliminações de patologias.
Analisa-se:
Anomalia congênita
Ossos calvária (presença ou ausência óssea)
Deformidade crânio (presente ou ausente)
Forma deformidades crânio (tipificação e relação entre patologias)
Acalvaria não é relacionada com a infecção, não há estudo
evidenciado em amostragem significativa que possa estabelecer relação com
etiologia infecciosa.
Herança genética e contágio não se relacionam:
Contágio não tem nada a ver com genética ou doenças herdadas dos
pais. Herança genética e contágio não são a mesma coisa.
Você nasce com seus genes em seu DNA, e eles não podem ser
transferidos.
Condições genéticas são herdadas dos pais, sendo incorporadas até
o nascimento. Não se pode adquirir uma condição genética após o nascimento.
Contagiosidade refere-se à transferência de um agente infeccioso
(por exemplo, vírus ou bactérias) entre as pessoas.
Diagnóstico errôneo de Acalvaria
Acalvaria pode ser diagnosticada como uma encefalocele por ultrassom
pré-natal, considerando a incidência de ambos, particularidades similares, e a
inviabilidade de exames específicos.
Mortes
Acalvaria é normalmente fatal, embora tenha sido relatada a
sobrevivência.
Índices estatísticos:
Prevalência: numero que geralmente se refere à população estimada
de pessoas que estão a gerir Acalvaria, num determinado período. Estes
resultados não se mostram como fidedignos, porque não se tem garantida a
precisão diagnóstica veste similaridades de outras patologias frente à acalvaria.
Incidência: refere-se à taxa anual de diagnóstico, ou o número de
novos casos de Acalvaria diagnosticados todos os anos. Assim, estes dois
tipos de estatísticas podem diferir: uma doença de curta duração, como a
gripe pode ter incidência anual elevado, mas prevalência baixa, mas uma
doença ao longo da vida, como o diabetes tem uma baixa incidência
anual, mas alta prevalência.
Acalvaria é listada como uma “doença rara" pelo Instituto de
Doenças Raras (ORD) dos Institutos Nacionais de Saúde (NIH). Isto
significa que Acalvaria, ou um subtipo de Acalvaria, afeta menos de 200.000
pessoas na população dos EUA.
Orphanet,
que é um consórcio de parceiros europeus, atualmente define uma doença como
rara quando se atinge uma pessoa, por 2.000. Eles lista Acalvaria como
uma "doença
rara“. .
Ao clicar nos
links abaixo, poderá obter maiores informações sobre tais temas, contudo, habilite
tradutor em seu navegador.
Condições
ósseas: Condições que afetam os ossos
Condições
congênitas - defeitos físicos: congênitas (nascimento) defeitos, causando
um defeito estrutura física (em vez de metabólica).
Condições de
cabeça: Condições que afetam a cabeça
Condições
musculoesqueléticas: condições médicas que afetam o sistema musculoesquelético
de ossos, músculos e estruturas relacionadas.
Deformidade crânio:
Uma condição que é caracterizada por uma deformação que está localizado no
crânio
quarta-feira, 26 de setembro de 2012
segunda-feira, 24 de setembro de 2012
Síndrome Abruzzo Erickson
Síndrome Abruzzo Erickson , comumente conhecida como síndrome de CHARGE, é uma doença genética rara. O nome CHARGE na verdade um acrônimo para ajudar a descrever os sintomas mais comuns que podem ser vistos em crianças nascidas com essa condição.
Os sintomas

Os sintomas mais comuns de Síndrome Abruzzo Erickson incluem anomalias do sistema nervoso central, incluindo Colabamento do olho, vários tipos de defeitos cardíacos, retardo do crescimento, resultando em a pessoa ser pequena em estatura, atresia das coanas, anormalidades do ouvido, às vezes incluindo surdez. Muito poucas pessoas acometidas por esta síndrome apresentam todos estes sintomas, mas normalmente, pelo menos, dois ou três estão presentes na maioria dos casos.
O tratamento comum da orelha de alguém com Síndrome Abruzzo Erickson , é um implante para ajudar com a audição.
Causas
Este síndrome é causada por mutações do gene CHD7 que faz parte do cromossoma 8, na maioria dos pacientes.
Tratamentos
Crianças com síndrome de Abruzzo-Erickson pode sobreviver até a idade adulta e viver uma vida feliz e produtiva, desde que eles sejam tratados a partir de uma idade precoce. Essas crianças têm muitos problemas de risco de vida, que devem ser abordadas o mais precocemente possível. Avanços da medicina nas últimas décadas, tornaram muito mais comum as pessoas nascidas com síndrome de Abruzzo-Erickson viverem uma vida longa e feliz. A maioria das pessoas com essa síndrome têm problemas de audição, que é um dos primeiros sintomas, sendo fundamental na direção a seguir ao planejar um plano de tratamento ao longo da vida.
Não há cura para esta doença e grande parte dos tratamentos serão focados em abordar sintomas específicos, em vez de a própria síndrome. Coisas como fisioterapia e terapia ocupacional são importantes. Uma coisa importante a lembrar é que muitas crianças com essa síndrome são de inteligência normal, mas devido à sua deficiência pode precisar de ajuda especial. Alguns pais, subestimam a inteligência de seus filhos por causa dos problemas de audição, e outras questões que fazem a educação mais difícil.
Trabalhando com médicos para garantir um plano para que eles possam viver a vida como normal e a mais produtiva possível, é importante desde cedo. Na maioria das vezes, com especial atenção, eles podem ter uma vida muito longa e feliz. Monitorar o coração para os defeitos que são comuns, também é importante uma vez que existem medicamentos que podem ajudar a minimizar este risco, ou cirurgia para corrigir algumas das condições que são possíveis.
Diagnóstico
O diagnóstico da doença é tipicamente feito por médicos, observando vários dos sintomas mais comuns no paciente. Existem testes genéticos que podem ajudar a confirmar a condição, mas estes são caros para recorrer e, só tem uma taxa de 60% de sucesso sendo que, na maioria dos casos um diagnóstico confirmado também não altera as opções de tratamento, traduzindo que há pouca vantagem em procurar os testes genéticos, na maioria dos casos.
domingo, 23 de setembro de 2012
Adrenoleucodistrofia
A adrenoleucodistrofia, também conhecida pelo acrônimo ALD, é uma doença genética rara, incluída no grupo das leucodistrofias, e que tem duas formas, sendo a mais comum à forma ligada ao cromossomo X, sendo uma herança ligada ao sexo de caráter recessivo, transmitida por mulheres portadoras e que afeta fundamentalmente homens.
O filme Lorenzo's Oil ("O óleo de Lorenzo") trata da manifestação da doença e da busca pela cura por parte dos pais de Lorenzo Odone, menino portador de ALD, sendo baseado em fatos reais.
Como ocorre
Na ALD, a atividade anormal dos peroxissomos leva a um acúmulo excessivo de ácidos graxos de cadeia muito longa (AGCML) constituídos de 24 ou 26 átomos de carbono em tecidos corporais, sobretudo no cérebro e nas glândulas adrenais. A consequência desse acúmulo é a destruição da bainha de mielina, o revestimento dos axônios das células nervosas, afetando, assim, a transmissão de impulsos nervosos.
O gene defeituoso que ocasiona a doença está localizado no lócus Xq-28 do cromossomo X. Tal gene é responsável pela codificação de uma enzima denominada ligase acil CoA gordurosa, que é encontrada na membrana dos peroxissomos e está relacionada ao transporte de ácidos graxos para o interior dessa estrutura celular. Como o gene defeituoso ocasiona uma mutação nessa enzima, os AGCML ficam impedidos de penetrar nos peroxissomos e se acumulam no interior celular. Os mecanismos precisos através dos quais os AGCML ocasionam a destruição da bainha de mielina ainda são desconhecidos.
Constitui distúrbio hereditário do metabolismo dos ácidos graxos que afeta as glândulas adrenais, o sistema nervoso e também os testículos.
A incidência de ALD é de cerca de 1 para cada 10.000 indivíduos. As possibilidades de descendência a partir de uma mulher portadora da ALD e homem normal são:
25% de chances de nascer um filho normal;
25% de chances de nascer um filho afetado;
25% de chances de nascer uma filha normal;
25% de chances de nascer uma filha portadora heterozigota.
As chances de descendência para um homem afetado e mulher homozigota dominante, por sua vez, são:
Se tiver filhas, serão todas portadoras do gene, porém normais;
Se tiver filhos, serão todos normais;
Formas básicas da doença
Neonatal
Manifesta-se nos primeiros meses de vida. Os genes anormais que causam a forma neonatal da ALD não estão localizados no cromossomo X, o que significa que pode afetar tanto meninos quanto meninas.
Período de sobrevida: 5 anos.
Sintomas: Retardo; disfunção adrenal; deterioração neurológica; degeneração retinal; convulsões; hipertrofia do fígado; anomalias faciais; músculos fracos.
Clássica ou infantil
Forma mais grave da ALD, desenvolvida por cerca de 35% dos portadores da doença. Manifesta-se no período de 4 a 10 anos de idade.
Período de sobrevida: 10 anos.
Sintomas: Problemas de percepção; disfunção adrenal; perda da memória, da visão, da audição, da fala; deficiência de movimentos de marcha; demência grave.
Adulta (AMN)
Forma mais leve que a clássica. Manifesta-se no início da adolescência ou no início da idade adulta.
Período de sobrevida: Décadas.
Sintomas: Dificuldade de deambulação; disfunção adrenal; incontinência urinária; deterioração neurológica.
ALD em mulheres
Embora a doença se manifeste principalmente em homens, mulheres portadoras também podem desenvolver uma forma leve da ALD, com sintomas como ataxia e fraqueza ou paralisação dos membros inferiores.
Sintomas predominantes na forma Clássica ou infantil
Hiperatividade
Diminuição do desempenho escolar
Diminuição da compreensão da comunicação verbal (afasia)
Pode haver sinais de função adrenal anormal (insuficiência adrenal).
Sintomas gerais de insuficiência adrenal = fraqueza muscular, emaciação (perda de peso e de massa muscular),
Diminuição do apetite (anorexia),
Aumento da pigmentação cutânea (escurecimento da pele),
Deterioração do controle motor fino.
Outros sintomas complementares:
Olhos vesgos (estrabismo convergente);
Deterioração da escrita (incapacidade de articular frases ou palavras antes comuns);
Dificuldade de deglutição (disfagia intensa);
Alterações no tônus muscular, especialmente espasmos musculares e espasticidade (contrações involuntárias);
Deterioração progressiva do sistema nervoso:
Convulsões;
Paralisia (inicia com dificuldade de controle de marcha, miastenia difusa, atrofia de extremidades “mãos fechadas”);
Perda da audição;
Deficiência visual ou cegueira;
Estado vegetativo (total incapacidade de auto cuidado e interação);
Crise: choque;
Cianose (extremidades e lábios de cores roxas azuladas);
Pulso rápido e fraco (taquicardia Filiforme);
Pele fria (hipotermia sensorial);
Respiração rápida (taquipnéia).
Tratamento
Pacientes com ALD devem ser testados periodicamente para a função das glândulas adrenais. O tratamento com hormônios das glândulas adrenais é indicado para salvar vidas. Os tratamentos dos sintomas da ALD incluem fisioterapia, apoio psicológico e educação especial. Há evidências de que o tratamento pelo óleo de Lorenzo pode adiar reduzir ou adiar os efeitos da ALD em meninos afetados pela variante genética associada ao cromossoma X. Transplantes de medula trazem benefícios a longo prazo quando os efeitos começam a se manifestar, mas a cirurgia tem altos riscos de mortalidade e morbidade, e não é recomendada para quem tem efeitos severos, ou para as formas que se manifestam em adultos ou de forma neonatal. A administração oral do ácido docosaexaenoico (DHA) pode ajudar crianças com a forma de ALD neonatal.
O médico deve solicitar os exames abaixo:
-A ressonância magnética do crânio que mostra os danos causados na substância branca do cérebro;
- A química sérica. Ela indica a quantidade elevada de ácidos graxos de cadeia longa.
-Uma biopsia de pele e cultura de fibroblastos indicando os níveis elevados de ácidos graxos de cadeia longa.
-Ultrassom das glândulas suprarrenais e testículos.
O tratamento da disfunção adrenal é feita com esteroides suplementares como hidrocortisona e cortisol.
Até o momento, não existe um tratamento específico disponível para a adrenoleucodistrofia.
Uma dieta baixa em ácidos graxos de cadeia longa e a administração de óleos especiais têm se mostrado capazes de reduzir os níveis plasmáticos dos ácidos graxos de cadeia longa. Esse tipo de óleo é conhecido como Óleo de Lorenzo, em homenagem ao filho do casal que descobriu o tratamento. A eficácia dessa dieta no tratamento da adrenoleucodistrofia ainda está sendo avaliada.
Expectativas (prognóstico):
A forma infantil da adrenoleucodistrofia é uma doença progressiva que leva a uma inabilidade profunda.
As formas tardias são muito menos perigosas.
Referências:
National Institutes of Health, National Institute of Neurological Disorders and Stroke, NINDS Adrenoleukodystrophy Information Page [em linha]
Página sobre a adrenoleucodistrofia no site lookfordiagnosis.com
Artigo sobre tratamento promissor através de um ácido (SAHA) que normalizou os ácidos graxos de cadeia longa e as inflamações secundarias. (em inglês)
http://gabrielpollaco.blogspot.com.br
Síndrome de Aase
A síndrome de Aase é uma
doença hereditária rara, caracterizada pela presença de anemia hipoplásica e do
esqueleto. Aparece mais frequentemente em mulheres, os sintomas da doença
podem ser detectados em recém-nascidos, ou na primeira infância. A
etiologia (estudo das causas da doença), foi descrita pela primeira vez em
1969 por Jon M. Aase e David W. Smith.
Clinicamente caracterizada pela
presença de polegares com três falanges, acompanhada por sintomas de pele, mucosas
pálidas por anemia hipoplásica (diminuição de células vermelhas do sangue, que afetam
e produzem falhas em todos os elementos figurados do sangue, determinam como
resultado a medula óssea não pode gerar novas células), devido à hipoplasia
(desenvolvimento incompleto ou com defeito) da medula óssea e, em casos muito
graves de anemia, letargia (anormal e sono profundo), irritabilidade e falha
(falha funcional) do coração. Foi
acompanhada (líquido no cérebro) hidrocefalia, contraturas articulares, o
fechamento tardio das fontanelas, orelhas malformadas e palato (fechamento
incompleto do palato duro), escoliose (curvatura anormal da coluna oblíqua),
luxação (deslocamento uma má posição conjunta) do quadril, dos pés, testa,
blefarofimose (fissura palpebral pequena), ampla ponte nasal, dermatoglifia
(padrões formados pelas cordilheiras e sulcos das mãos e pés) ligeiramente
marcado, Dandy Walker anomalia (acúmulo de hidrocefalia, o fluido no cérebro
por atresia, leva a oclusão da abertura natural, resultante em buracos
congênitos anômalos Magendi e Luschka), micrognatia (mandíbula anormalmente
pequena) e ptose palpebral (pálpebras caídas).
Menor frequência pode ser
acompanhada por defeito do septo (comunicação anormal entre os ventrículos do
coração), neuroblastoma (tumor maligno das células nervosas embrionárias),
estrabismo (olhos cruzados de um de sua direção normal, de modo que os eixos
visuais não podem ser dirigidos ao mesmo tempo retardo de crescimento no mesmo
ponto), surdez, centro de ossificação do esterno e apenas moderada. Diagnóstico
é suspeita clínica, confirmada por biópsia (cirurgia que envolve a remoção do
órgão fragmento de vida individual ou tumor, a fim de apresentar para exame
microscópico) com medula óssea de células vermelhas hipoplasia caracterizado
por déficit de precursores de eritrócitos com um motivo mielócitos /
eritrócitos baixo.
 O diagnóstico é obtido por meio
de:
O diagnóstico é obtido por meio
de:
Biópsia da medula óssea
(mielograma);
Ecocardiograma pode
detectar anormalidades cardíacas, defeitos do septo ventricular (coração parede
que separa o ventrículo esquerdo do direito) e,
Estudo radiológico identifica
alterações esqueléticas.
Hemograma, que identifica a presença
de anemia e uma redução na contagem de glóbulos brancos;
Não existe um tratamento
específico para a doença, o objetivo do tratamento é controlar a anemia
hipoplásica, que são em muitas vezes utilizados esteroides e transfusões
repetidas.
Para o tratamento desta
patologia, costuma-se realizar diversas transfusões sanguíneas no primeiro ano
de vida para tratar a anemia. A prednisona pode ser utilizada, porém não é
recomendada durante a infância, pois apresenta efeitos colaterais sobre o
crescimento e o desenvolvimento do cérebro. Também existe a opção de um
transplante de medula, caso as outras opções de tratamento não levem a
resultados satisfatórios.
Quelantes de ferro deve ser usado
para evitar a hemossiderose (depósito de um pigmento amarelo contendo ferro nos
tecidos), secundária a múltiplas transfusões.
Complicações são os resultantes
de anemia, principalmente esplenomegalia (baço anormalmente grande), causada
por hemossiderose (transfusões repetidas e efeitos colaterais dos esteroides),
como retardo de crescimento, osteoporose (desmineralização esquelética
generalizada), edema (acúmulo de líquido em excesso no seroalbuminoso tecido
celular), hipertensão, diabetes, úlcera gástrica, catarata e glaucoma.
Em casos refratários deve ser
considerada a possibilidade de transplante de medula óssea. Alguns autores
consideram a síndrome de Aase, herdada como traço genético autossômico
dominante.
A base genética dessa patologia
não é conhecida até o momento. A anemia da síndrome resulta de um
desenvolvimento retardado da medula óssea, local onde as células
sanguíneas são produzidas.
Referências:
Instituto de Pesquisa de Doenças Raras: http://iier.isciii.es/
sábado, 22 de setembro de 2012
Síndrome de Aarskog-Scott, Distúrbio de Hipertelorismo.
Nomes alternativos:
Síndrome de Aarskog-Scott, Distúrbio
de Hipertelorismo.
Definição:
Doença hereditária que se caracteriza
por baixa estatura, anormalidades faciais e genitais.
 A Síndrome de Aarskog é uma
enfermidade genética rara, recessiva, ligada ao cromossomo X. Ela se
caracteriza por um aumento visível da separação entre olhos, dedos curtos
(braquidactilia), face arredondada, baixa estatura, pálpebras caídas, nariz
pequeno e alargado, anomalias labiais, palatais (fissuras) e do pavilhão
auricular, estrabismo, astigmatismo, dentição atrasada, aumento da
flexibilidade de falanges, prega simiesca da palma da mão, pés pequenos e
chatos, hérnias e outras anomalias.
A Síndrome de Aarskog é uma
enfermidade genética rara, recessiva, ligada ao cromossomo X. Ela se
caracteriza por um aumento visível da separação entre olhos, dedos curtos
(braquidactilia), face arredondada, baixa estatura, pálpebras caídas, nariz
pequeno e alargado, anomalias labiais, palatais (fissuras) e do pavilhão
auricular, estrabismo, astigmatismo, dentição atrasada, aumento da
flexibilidade de falanges, prega simiesca da palma da mão, pés pequenos e
chatos, hérnias e outras anomalias.
Ocasionalmente apresenta escoliose,
espinha bífida e retardo mental leve. Não se conhece bem as causas da Síndrome
de Aarskog. Pesquisadores acham que essa desordem pode estar associada a um
componente genético ligado ao cromossomo X.
Causas, incidência e fatores de
risco:
 A síndrome de Aarskog, ligada ao
cromossomo X, parece ser um distúrbio hereditário autossômico recessivo ou semi
dominante. Este distúrbio afeta principalmente as pessoas do sexo masculino,
embora as do sexo feminino possam ser portadoras de algumas dessas
características.
A síndrome de Aarskog, ligada ao
cromossomo X, parece ser um distúrbio hereditário autossômico recessivo ou semi
dominante. Este distúrbio afeta principalmente as pessoas do sexo masculino,
embora as do sexo feminino possam ser portadoras de algumas dessas
características.
Observação:
A síndrome de Aarskog é uma
doença genética rara, existem apenas 100 casos relatados no mundo.
Os seus efeitos incluem baixa
estatura, anomalias faciais, genitais e musculoesqueléticas.
Está ligada ao cromossoma X e é de
carácter recessivo. Afeta, sobretudo indivíduos do sexo masculino. É também
conhecida como síndrome de Aarskog-Scott, displasia
faciogenital ou síndrome faciodigitogenital.
Sintomas:
· baixa estatura, característica que pode não ser evidente até a criança
ter entre 1 e 3 anos de idade.
· atraso da maturação sexual
· face arredondada
· linha capilar formando um bico na testa
· olhos muito separados, com as pálpebras caídas.
· nariz pequeno com as narinas projetadas para frente
· parte média da face pouco desenvolvida
· fenda larga sobre o lábio superior e dobra abaixo do lábio inferior
· atraso na erupção dos dentes
· parte superior do pavilhão auricular ligeiramente dobrada
· mãos e pés pequenos e largos
· dedos das mãos e dos pés curtos, interligados por fina membrana.
· prega simiesca (única) na palma da mão
· esterno ligeiramente côncavo
· umbigo protuberante
· hérnias inguinais
· escroto "vazio", testículos retidos.
· deficiência mental leve
· peito escavado
Sinais e exames:
Raios-X para detecção de
anormalidades esqueléticas.
Tratamento:
Pode-se tentar um tratamento
ortodôntico para algumas anormalidades faciais. A utilização de hormônio do
crescimento não tem demonstrado eficácia no tratamento da baixa estatura,
característica deste distúrbio.
Expectativas (prognóstico):
Pode haver um leve atraso mental,
embora as crianças afetadas tenham, em geral, um bom desempenho social. Alguns
homens podem apresentar diminuição da fertilidade.
Complicações:
Achados recentes têm incluído
alterações císticas no cérebro e convulsões generalizadas.
Solicitação de assistência médica:
Solicite assistência médica se a
criança apresentar atraso no crescimento ou qualquer das anormalidades
descritas acima.
Referencias:
Assinar:
Comentários (Atom)
-
Em virtude de, estar acompanhando meu sobrinho em tratamento de saúde, as postagens estão adiadas por intervenção e força do momento. Grato...
-
 A Acanthosis nigricans (AN) é condição dermatológica caracterizada por espessamento, hiperpigmentação e acentuação das linhas da pele, ...
A Acanthosis nigricans (AN) é condição dermatológica caracterizada por espessamento, hiperpigmentação e acentuação das linhas da pele, ...